
Page 113 - 中国全科医学2022-05
P. 113
·622· http://www.chinagp.net E-mail:zgqkyx@chinagp.net.cn
反应可,全身皮肤黏膜未见出血点及瘀斑,毛发稍浓密。心
肺查体无明显异常,腹稍膨隆,触之软,肝脏肋下 2 cm,脾 患儿
脏肋下未触及。外生殖器及神经系统查体无异常。
9
实验室检查,血常规:WBC 6.09×10 /L,Hb 116 g/L,N
9
9
0.47×10 /L,PLT 326×10 /L;血生化:ALT 66 U/L,AST 194
U/L,肾功能、电解质大致正常;多次便常规可见脂肪滴;血
清免疫球蛋白、抗核抗体(ANA)、直接抗人球蛋白试验(DAT)、
间接抗人球蛋白试验(IAT)等均无异常;骨髓细胞形态学检
查未见明显异常;腹部 B 超提示肝脏轻度增大。
患儿父亲
为进一步明确诊断,经患儿监护人知情同意后取患儿及
其父母的外周血各 2 ml,乙二胺四乙酸(EDTA)抗凝,提取
基因组 DNA,针对 816 个遗传性血液系统疾病相关基因的外
显子和侧翼序列进行高通量测序。利用 Illumina NextSeq 500
第二代测序仪进行测序。本实验中样品基因的平均测序深度
约为 96×,捕获区覆盖度达 90% 以上,为找出致病性点突变,
参考 dbSNP 数据库、HapMap 数据库、千人基因组数据库及
ESP 数据库和内部正常对照人群数据库,将频率小于 0.05 的 患儿母亲
变异视为可疑,并进一步行 Sanger 测序验证(以上由北京迈
基诺基因科技股份有限公司提供技术支持)。测序结果发现:
3 例患儿均携带 SBDS 基因复合杂合突变。3 例患儿 SBDS 基
因突变位点相同,均有 2 个已知致病性杂合突变:c.258+2T>C
和c.184A>T。经家系验证分析,3例患儿父亲均存在c.258+2T>C
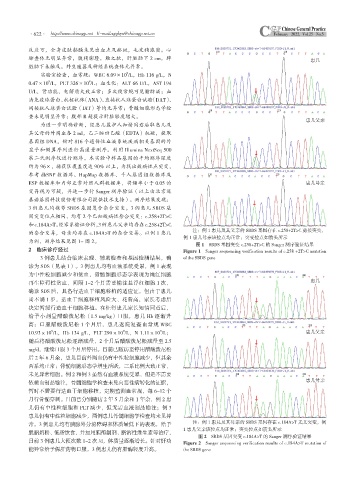
注:例 1 患儿及其父亲的 SBDS 基因存在 c.258+2T>C 剪接突变,
的杂合变异,母亲均存在 c.184A>T 的杂合变异。以例 1 患儿
例 1 患儿母亲该位点均正常;突变位点如箭头所示
为例,测序结果见图 1~ 图 2。
图 1 SBDS 基因突变 c.258+2T>C 的 Sanger 测序验证结果
2 临床诊疗经过 Figure 1 Sanger sequencing verification results of c.258 +2T>C mutation
3 例患儿结合临床表现、辅助检查和基因检测结果,确 of the SBDS gene
诊为 SDS(见表 1)。3 例患儿均有血液系统受累,例 1 表现
为中性粒细胞减少和贫血,骨髓细胞形态学表现为纯红细胞
再生障碍性贫血,间隔 1~2 个月需要输注悬浮红细胞 1 次, 患儿
确诊 SDS 后,具备行造血干细胞移植的适应证,但由于患儿
尚不满 1 岁,造血干细胞移植风险大、花费高,家长考虑后
决定暂缓行造血干细胞移植。在征得患儿家长知情同意后,
给予小剂量醋酸泼尼松(1.5 mg/kg)口服,患儿 Hb 逐渐升
高;口服醋酸泼尼松 1 个月后,患儿返院复查血常规 WBC
9
9
9
10.93×10 /L,Hb 134 g/L,PLT 290×10 /L,N 1.11×10 /L; 患儿父亲
随后将醋酸泼尼松逐渐减量,2 个月后醋酸泼尼松减量至 2.5
mg/d,继续口服 3 个月后停用。目前已随访至停用醋酸泼尼松
后 2 年 6 月余,患儿目前外周血仍有中性粒细胞减少,但其余
两系均正常;骨髓细胞形态学增生活跃,三系比例大致正常,
未见异常细胞。例 2 和例 3 虽然有血液系统受累,但是不需要
依赖血制品输注,骨髓细胞学检查未见向恶性病转化的证据, 患儿母亲
暂时不需要行造血干细胞移植,定期监测血常规,每 6~12 个
月行骨髓穿刺。目前已分别随访 2 年 5 月余和 1 年余,例 2 患
儿仍有中性粒细胞和 PLT 减少,但无需血液制品输注;例 3
患儿仍有中性粒细胞减少,两例患儿骨髓细胞学检查均未见异
常。3 例患儿均有胰腺外分泌障碍和体质量低下的表现,给予 注:例 1 患儿及其母亲的 SBDS 基因存在 c.184A>T 无义突变,例
1 患儿父亲该位点均正常;突变位点如箭头所示
脱脂奶粉、低脂饮食,并加用胰酶制剂、脂溶性维生素等治疗,
图 2 SBDS 基因突变 c.184A>T 的 Sanger 测序验证结果
目前 3 例患儿大便次数 1~2 次 /d,体质量逐渐增长。针对肝功 Figure 2 Sanger sequencing verification results of c.184A>T mutation of
能异常给予保肝药物口服,3 例患儿仍有肝酶轻度升高。 the SBDS gene