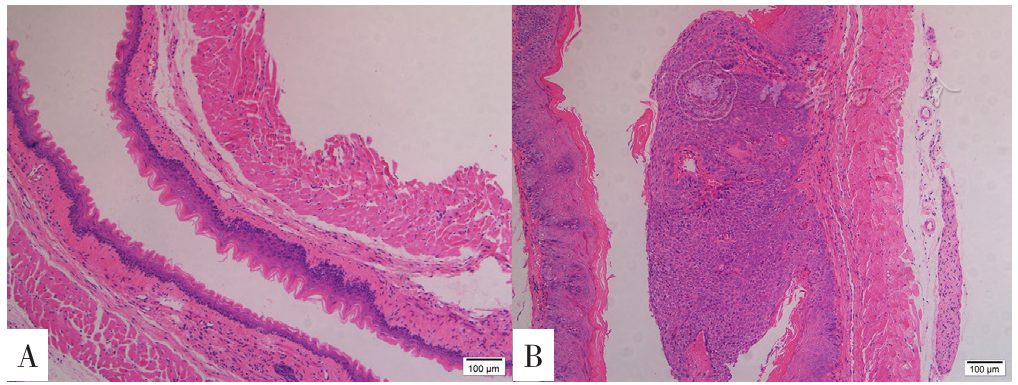
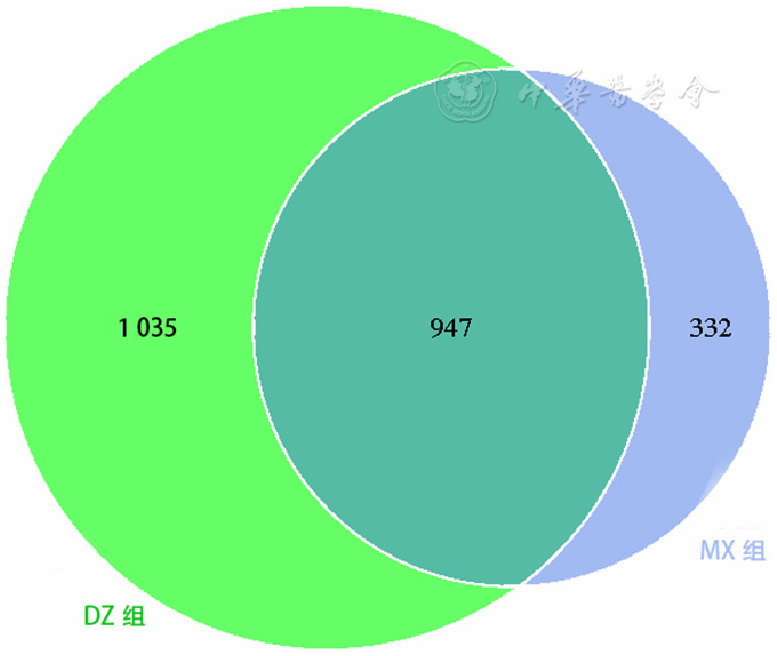
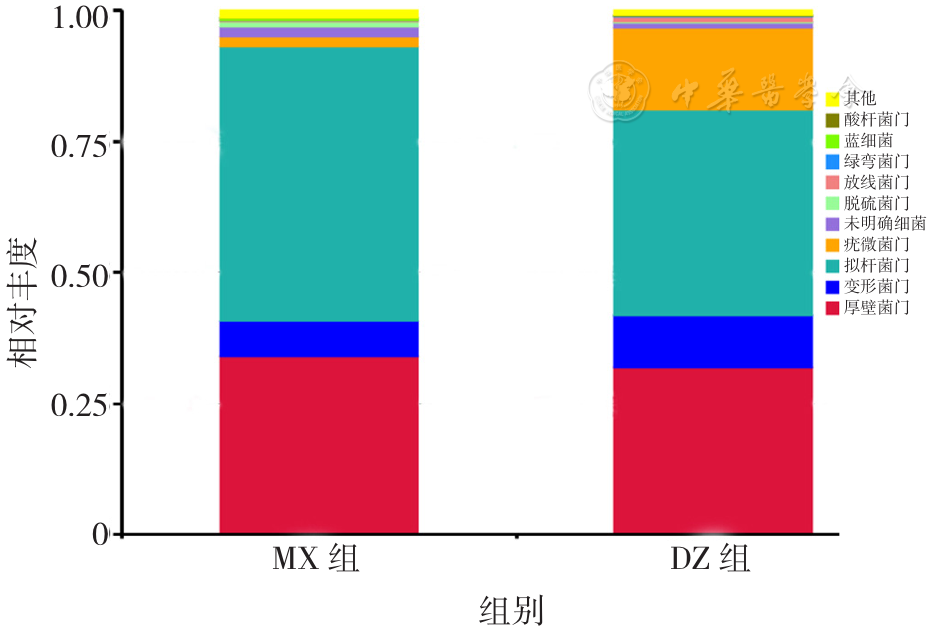
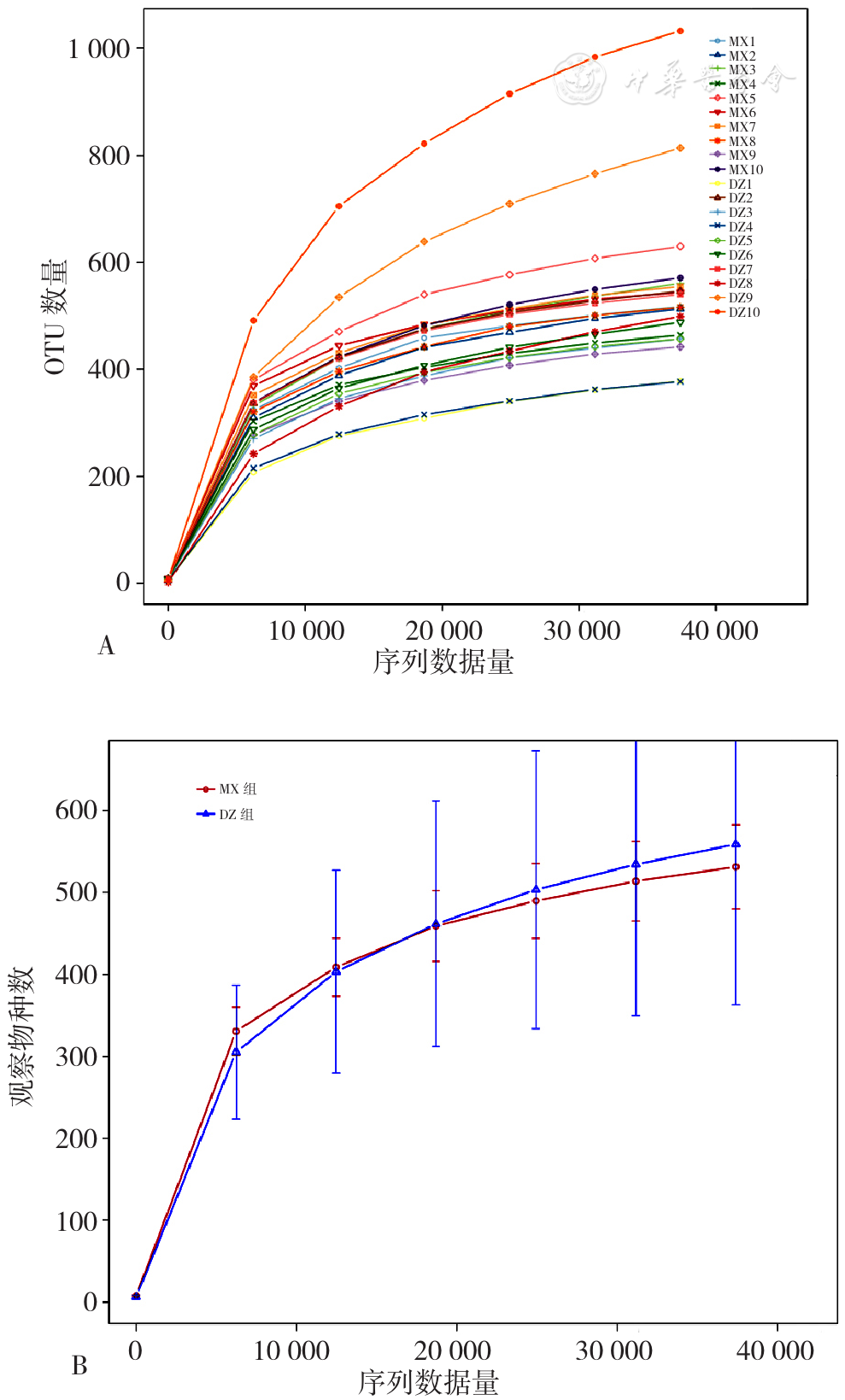
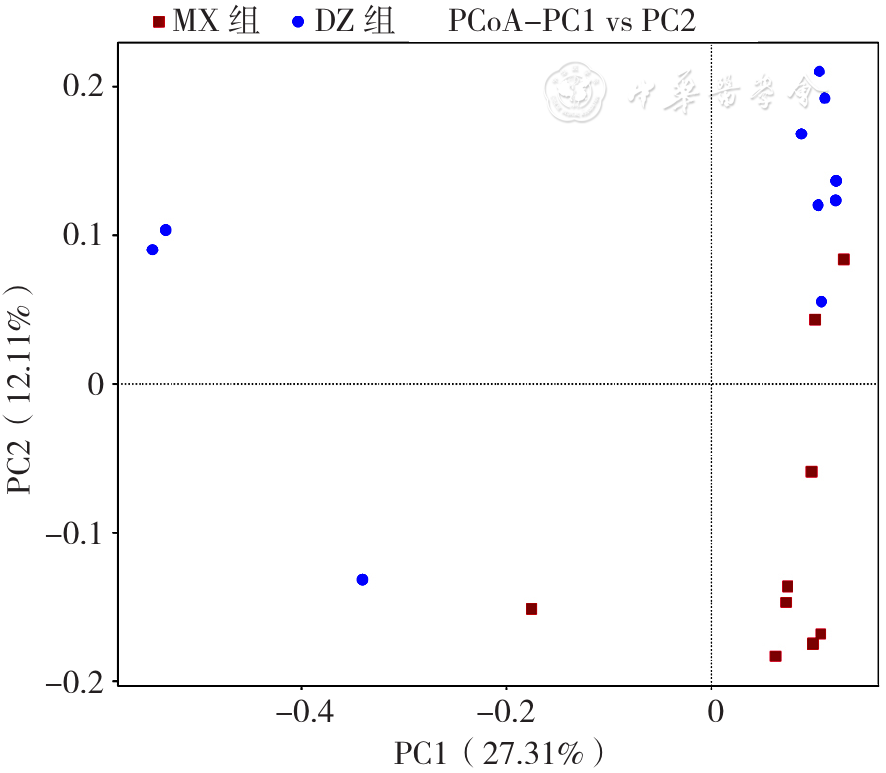
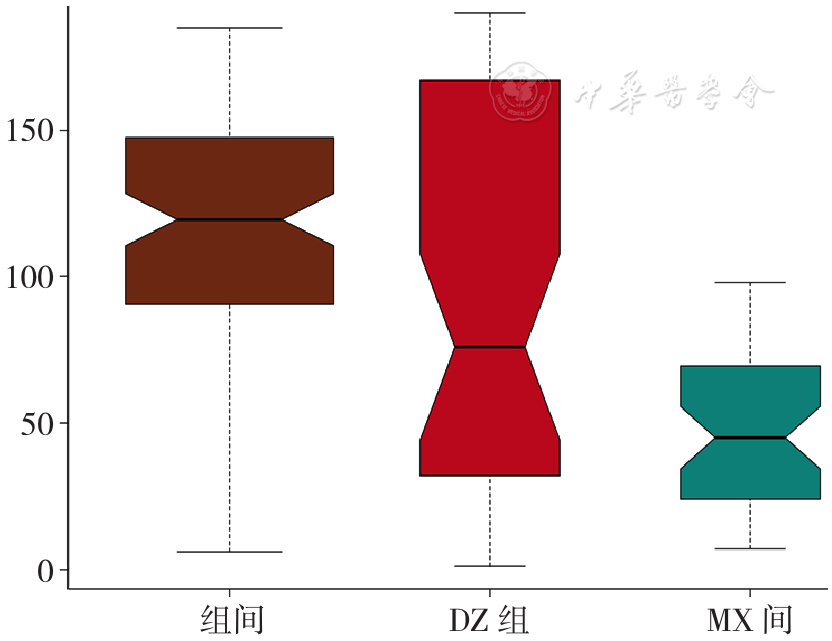
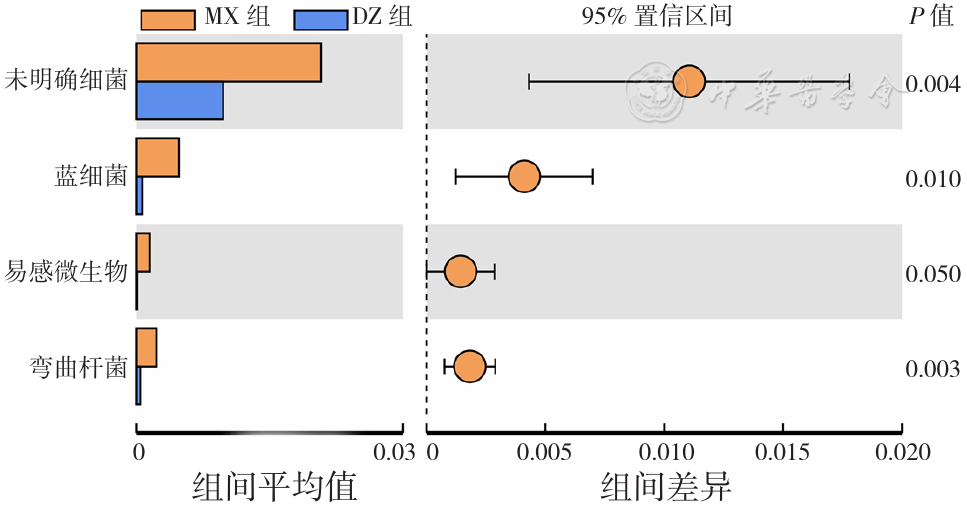
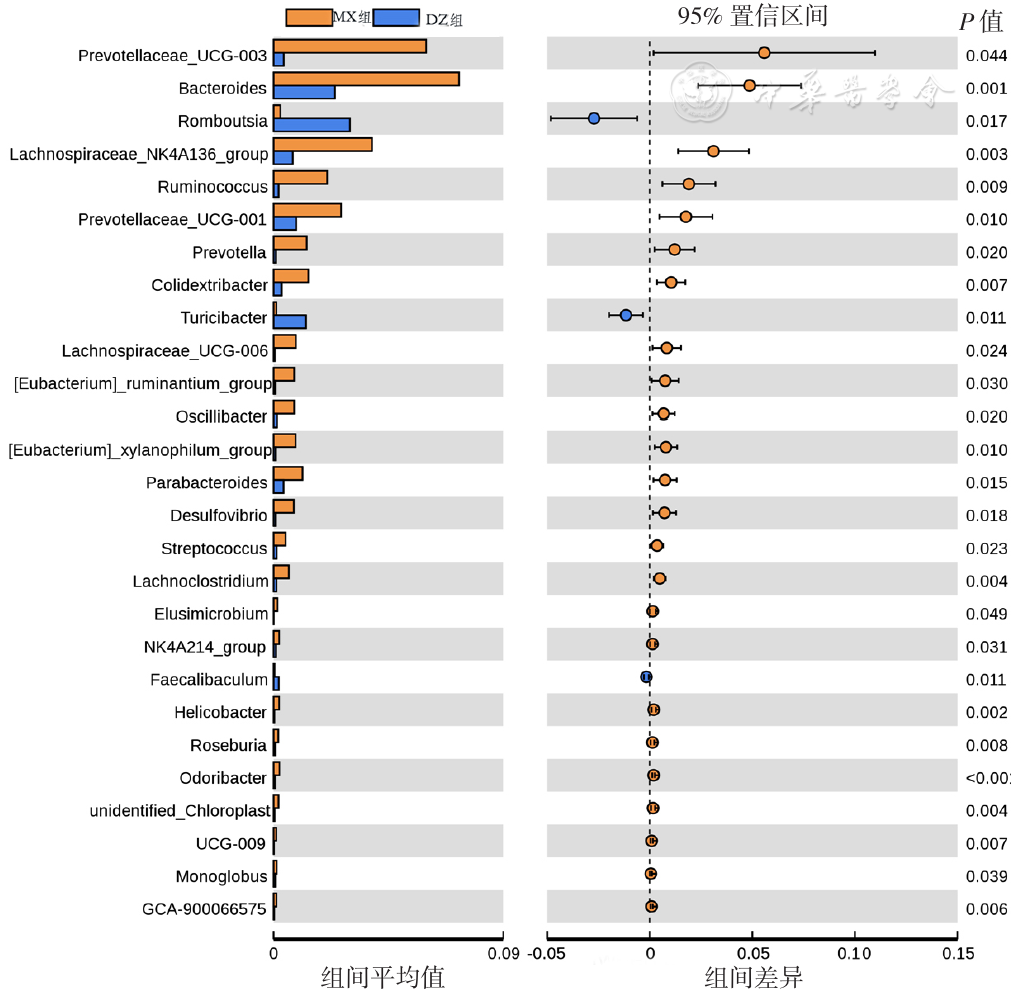
| [1] |
SUNG H, FERLAY J, SIEGEL R L,et al. Global cancer statistics 2020:GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin,2021,71(3):209-249. DOI:10.3322/caac.21660.
|
| [2] |
刘宗超,李哲轩,张阳,等. 2020全球癌症统计报告解读[J]. 肿瘤综合治疗电子杂志,2021,7(2):1-13. DOI:10.12151/JMCM.2021.02-01.
|
| [3] |
GAO S G, LI S G, MA Z K,et al. Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer[J]. Infect Agent Cancer,2016,11:3. DOI:10.1186/s13027-016-0049-x.
|
| [4] |
ZHAO Q F, YANG T, YAN Y F,et al. Alterations of oral microbiota in Chinese patients with esophageal cancer[J]. Front Cell Infect Microbiol,2020,10:541144. DOI:10.3389/fcimb.2020.541144.
|
| [5] |
王丽群,庞日朝,胡晓敏,等. 肠道菌群对色氨酸代谢的影响研究进展[J]. 中国比较医学杂志,2021,31(4):129-136. DOI:10.3969/j.issn.1671-7856.2021.04.019.
|
| [6] |
RAKOFF-NAHOUM S, FOSTER K R, COMSTOCK L E. The evolution of cooperation within the gut microbiota[J]. Nature,2016,533(7602):255-259. DOI:10.1038/nature17626.
|
| [7] |
YATSUNENKO T, REY F E, MANARY M J,et al. Human gut microbiome viewed across age and geography[J]. Nature,2012,486(7402):222-227. DOI:10.1038/nature11053.
|
| [8] |
MAYNARD C L, ELSON C O, HATTON R D,et al. Reciprocal interactions of the intestinal microbiota and immune system[J]. Nature,2012,489(7415):231-241. DOI:10.1038/nature11551.
|
| [9] |
WU Y L, CHEN Y N, LU Y F,et al. Structural features,interaction with the gut microbiota and anti-tumor activity of oligosaccharides[J]. RSC Adv,2020,10(28):16339-16348. DOI:10.1039/d0ra00344a.
|
| [10] |
LV J, JIA Y T, LI J,et al. Gegen Qinlian Decoction enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by remodelling the gut microbiota and the tumour microenvironment[J]. Cell Death Dis,2019,10(6):415. DOI:10.1038/s41419-019-1638-6.
|
| [11] |
BULLMAN S, PEDAMALLU C S, SICINSKA E,et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer[J]. Science,2017,358(6369):1443-1448. DOI:10.1126/science.aal5240.
|
| [12] |
ROUTY B, LE CHATELIER E, DEROSA L,et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors[J]. Science,2018,359(6371):91-97. DOI:10.1126/science.aan3706.
|
| [13] |
EUN C S, KIM B K, HAN D S,et al. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis,intestinal Metaplasia,and gastric cancer using pyrosequencing methods[J]. Helicobacter,2014,19(6):407-416. DOI:10.1111/hel.12145.
|
| [14] |
MA C, HAN M J, HEINRICH B,et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells[J]. Science,2018,360(6391):eaan5931. DOI:10.1126/science.aan5931.
|
| [15] |
GOPALAKRISHNAN V, SPENCER C N, NEZI L,et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients[J]. Science,2018,359(6371):97-103. DOI:10.1126/science.aan4236.
|
| [16] |
MATSON V, FESSLER J, BAO R Y,et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients[J]. Science,2018,359(6371):104-108. DOI:10.1126/science.aao3290.
|
| [17] |
COUGNOUX A, DALMASSO G, MARTINEZ R,et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype[J]. Gut,2014,63(12):1932-1942. DOI:10.1136/gutjnl-2013-305257.
|
| [18] |
CHENG W T, KANTILAL H K, DAVAMANI F. The mechanism of Bacteroides fragilis toxin contributes to colon cancer formation[J]. Malays J Med Sci,2020,27(4):9-21. DOI:10.21315/mjms2020.27.4.2.
|
| [19] |
SIVAN A, CORRALES L, HUBERT N,et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy[J]. Science,2015,350(6264):1084-1089. DOI:10.1126/science.aac4255.
|
| [20] |
位俊敏,李锐锋,杨冬阳,等. 食管鳞癌患者唾液菌群的研究[J]. 中山大学学报(医学科学版),2020,41(2):313-320. DOI:10.13471/j.cnki.j.sun.yat-sen.univ(med.sci).2020.0043.
|
| [21] |
DENG Y L, TANG D R, HOU P F,et al. Dysbiosis of gut microbiota in patients with esophageal cancer[J]. Microb Pathog,2021,150:104709. DOI:10.1016/j.micpath.2020.104709.
|
| [22] |
DEJEA C M, FATHI P, CRAIG J M,et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria[J]. Science,2018,359(6375):592-597. DOI:10.1126/science.aah3648.
|
| [23] |
KEKU T O, DULAL S, DEVEAUX A,et al. The gastrointestinal microbiota and colorectal cancer[J]. Am J Physiol Gastrointest Liver Physiol,2015,308(5):G351-363. DOI:10.1152/ajpgi.00360.2012.
|
| [24] |
WANG T T, CAI G X, QIU Y P,et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers[J]. ISME J,2012,6(2):320-329. DOI:10.1038/ismej.2011.109.
|
| [25] |
ZHANG Z C, CAO H Y, SONG N,et al. Long-term hexavalent chromium exposure facilitates colorectal cancer in mice associated with changes in gut microbiota composition[J]. Food Chem Toxicol,2020,138:111237. DOI:10.1016/j.fct.2020.111237.
|
| [26] |
SEOL M, LEE Y R, KIM K M,et al. The difference of the gut microbiota of gastric cancer in relation to Helicobacter pylori negativity and positivity[J]. J Clin Oncol,2019,37(4_):10. DOI:10.1200/jco.2019.37.4_suppl.10.
|