
Chinese General Practice ›› 2023, Vol. 26 ›› Issue (08): 939-950.DOI: 10.12114/j.issn.1007-9572.2022.0487
• Original Research • Previous Articles Next Articles
Received:2022-07-06
Revised:2022-08-28
Published:2023-03-15
Online:2022-11-30
Contact:
BIAN Yunfei
通讯作者:
边云飞
作者简介:基金资助:
Add to citation manager EndNote|Ris|BibTeX
URL: https://www.chinagp.net/EN/10.12114/j.issn.1007-9572.2022.0487
| 引物名称 | 引物序列 | 产物长度(bp) | 退火温度(℃) |
|---|---|---|---|
| H-BPI-S | GAATCAACTATGGTCTGGTGGCA | 106 | 60.62 |
| H-BPI-A | AAGGGAGGTGGATTGTGGTG | ||
| H-BIRC5-S | CGCATCTCTACATTCAAGAACTGG | 182 | 59.73 |
| H-BIRC5-A | GGTTTCCTTTGCATGGGGT | ||
| H-CXCL12-S | TGAGCTACAGATGCCCATGC | 137 | 60.18 |
| H-CXCL12-A | GCACACTTGTCTGTTGTTGTTCTTC | ||
| H-RNASE1-S | GCTCCTTCTGCTTGTCCTGAT | 149 | 60.07 |
| H-RNASE1-A | TCATCATTTGGTTACAGTAGGTGGA | ||
| H-F2R-S | CAGTGATTGGCAGTTTGGGTCT | 270 | 61.08 |
| H-F2R-A | TGACAGGTAGTGATGTTGAGCCC | ||
| H-ACTIN-S | CACCCAGCACAATGAAGATCAAGT | 317 | 61.60 |
| H-ACTIN-A | CCAGTTTTTAAATCCTGAGTCAAGC |
Table 1 Primer sequences for qRT-PCR
| 引物名称 | 引物序列 | 产物长度(bp) | 退火温度(℃) |
|---|---|---|---|
| H-BPI-S | GAATCAACTATGGTCTGGTGGCA | 106 | 60.62 |
| H-BPI-A | AAGGGAGGTGGATTGTGGTG | ||
| H-BIRC5-S | CGCATCTCTACATTCAAGAACTGG | 182 | 59.73 |
| H-BIRC5-A | GGTTTCCTTTGCATGGGGT | ||
| H-CXCL12-S | TGAGCTACAGATGCCCATGC | 137 | 60.18 |
| H-CXCL12-A | GCACACTTGTCTGTTGTTGTTCTTC | ||
| H-RNASE1-S | GCTCCTTCTGCTTGTCCTGAT | 149 | 60.07 |
| H-RNASE1-A | TCATCATTTGGTTACAGTAGGTGGA | ||
| H-F2R-S | CAGTGATTGGCAGTTTGGGTCT | 270 | 61.08 |
| H-F2R-A | TGACAGGTAGTGATGTTGAGCCC | ||
| H-ACTIN-S | CACCCAGCACAATGAAGATCAAGT | 317 | 61.60 |
| H-ACTIN-A | CCAGTTTTTAAATCCTGAGTCAAGC |
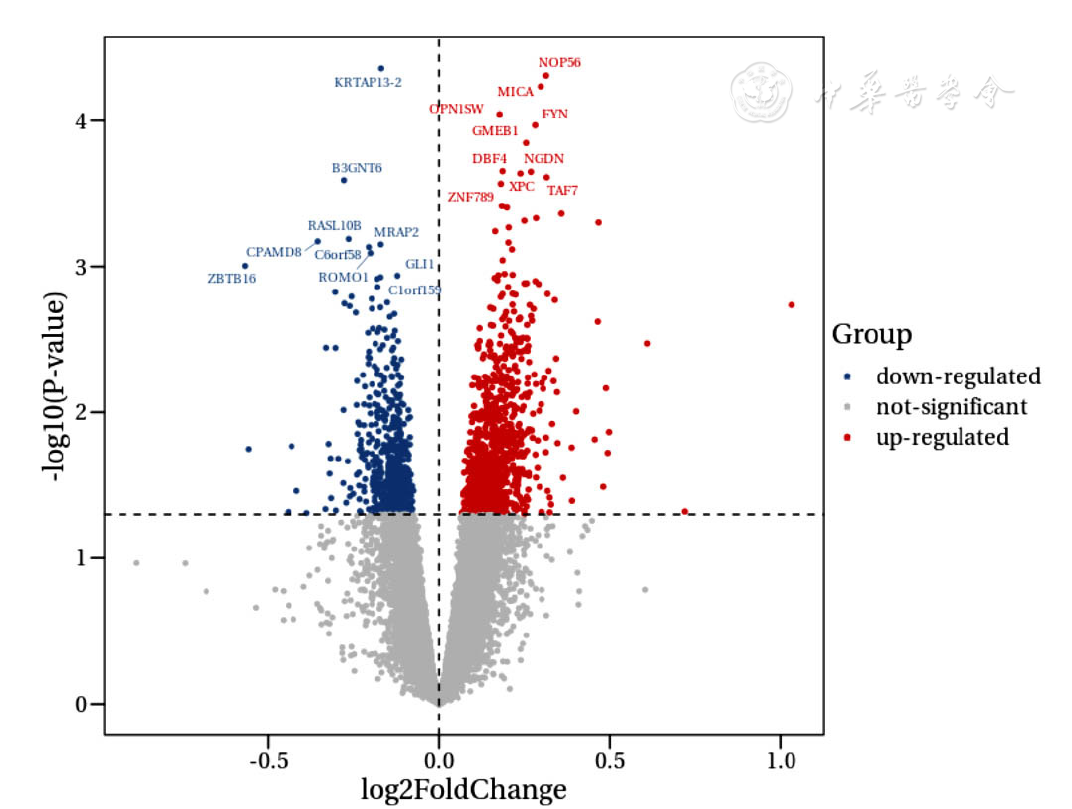
Figure 1 Volcano plot of differentially expressed genes in epicardial adipose tissue between CAD group and healthy control group
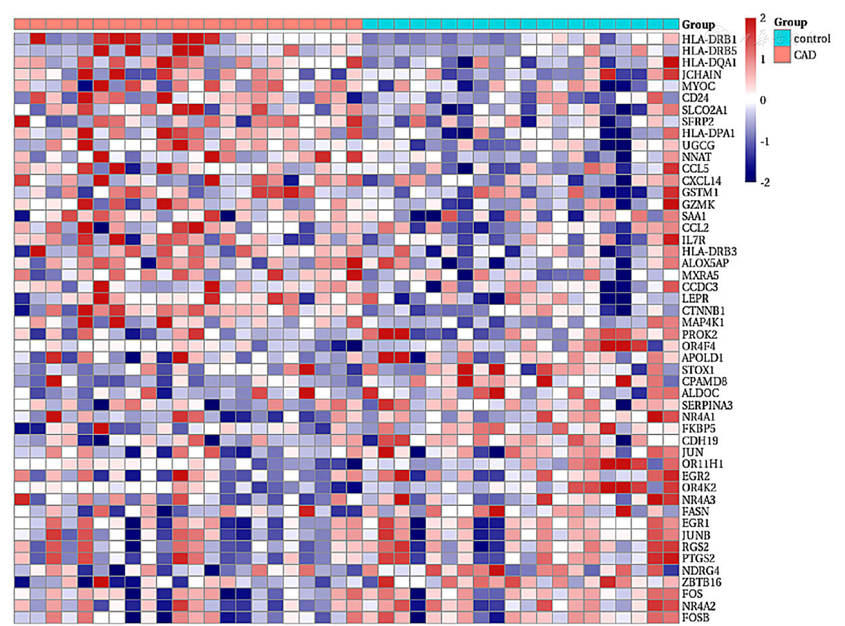
Figure 2 Heatmap of differentially expressed genes in epicardial adipose tissue between CAD group and healthy control group
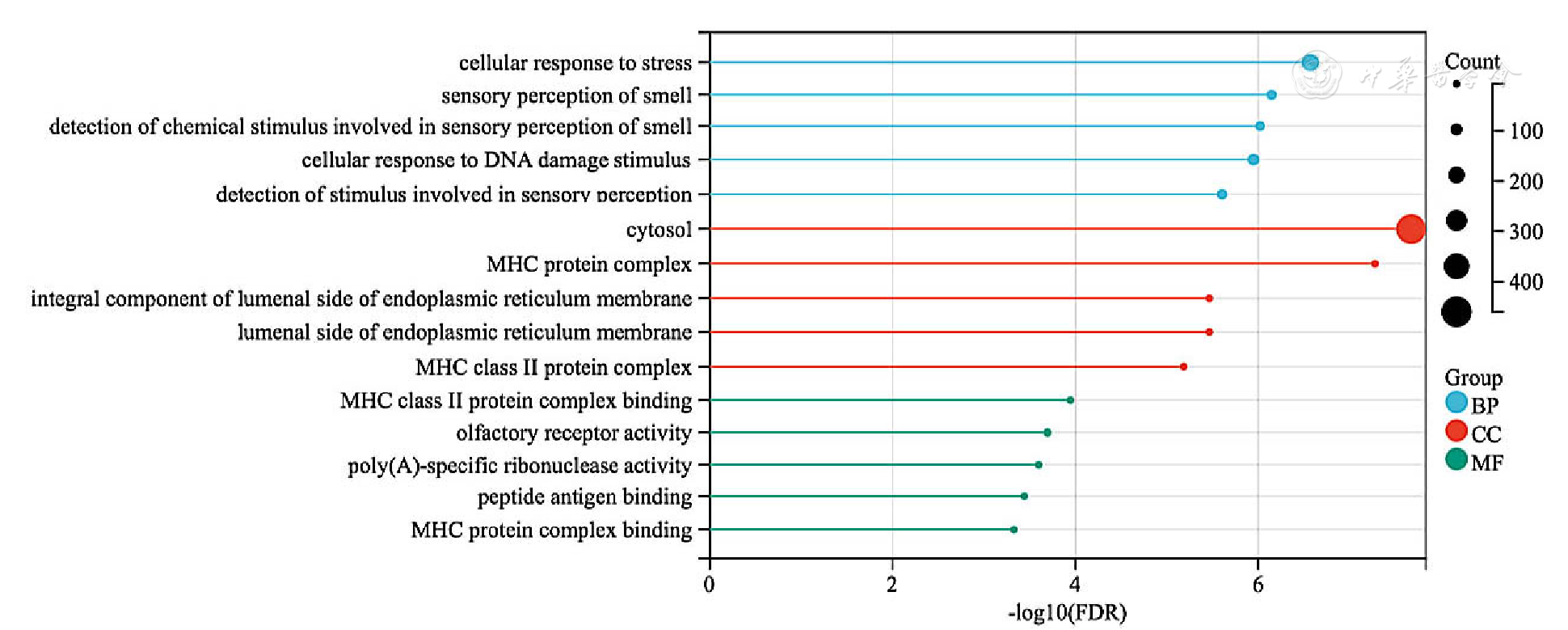
Figure 3 GO enrichment analysis of differentially expressed genes in epicardial adipose tissue
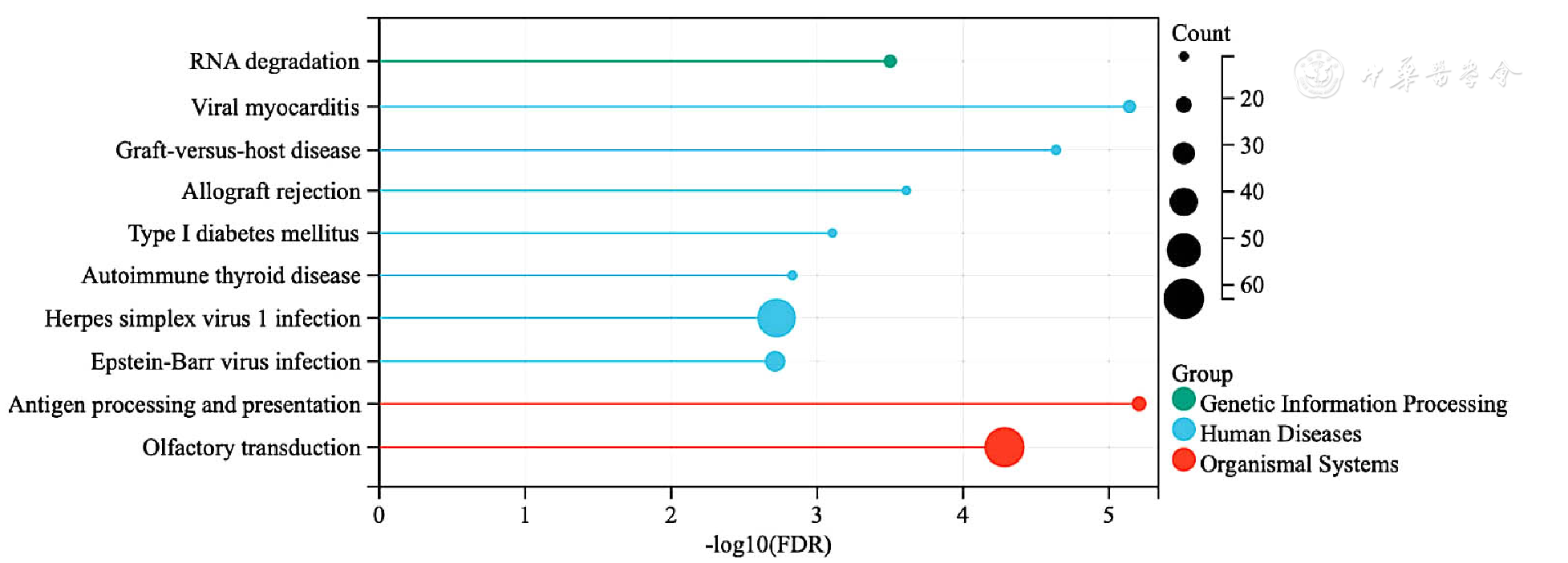
Figure 4 KEGG enrichment analysis of differentially expressed genes in epicardial adipose tissue
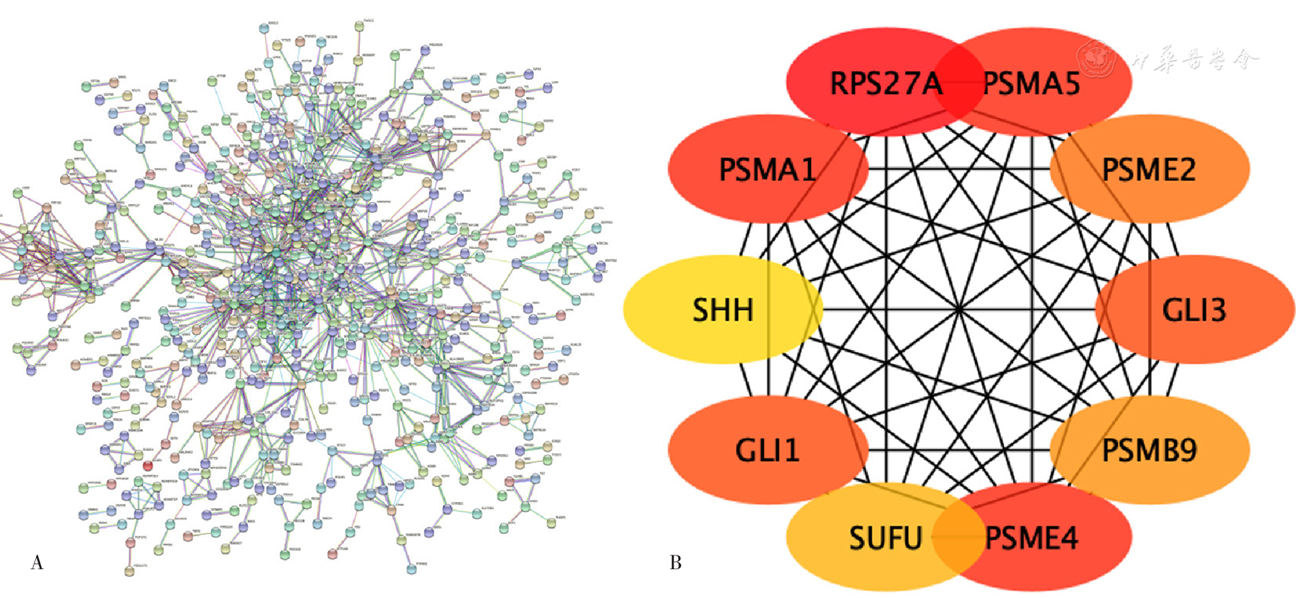
Figure 5 Differentially expressed genes in epicardial adipose tissue and the key genes identified in the PPI network
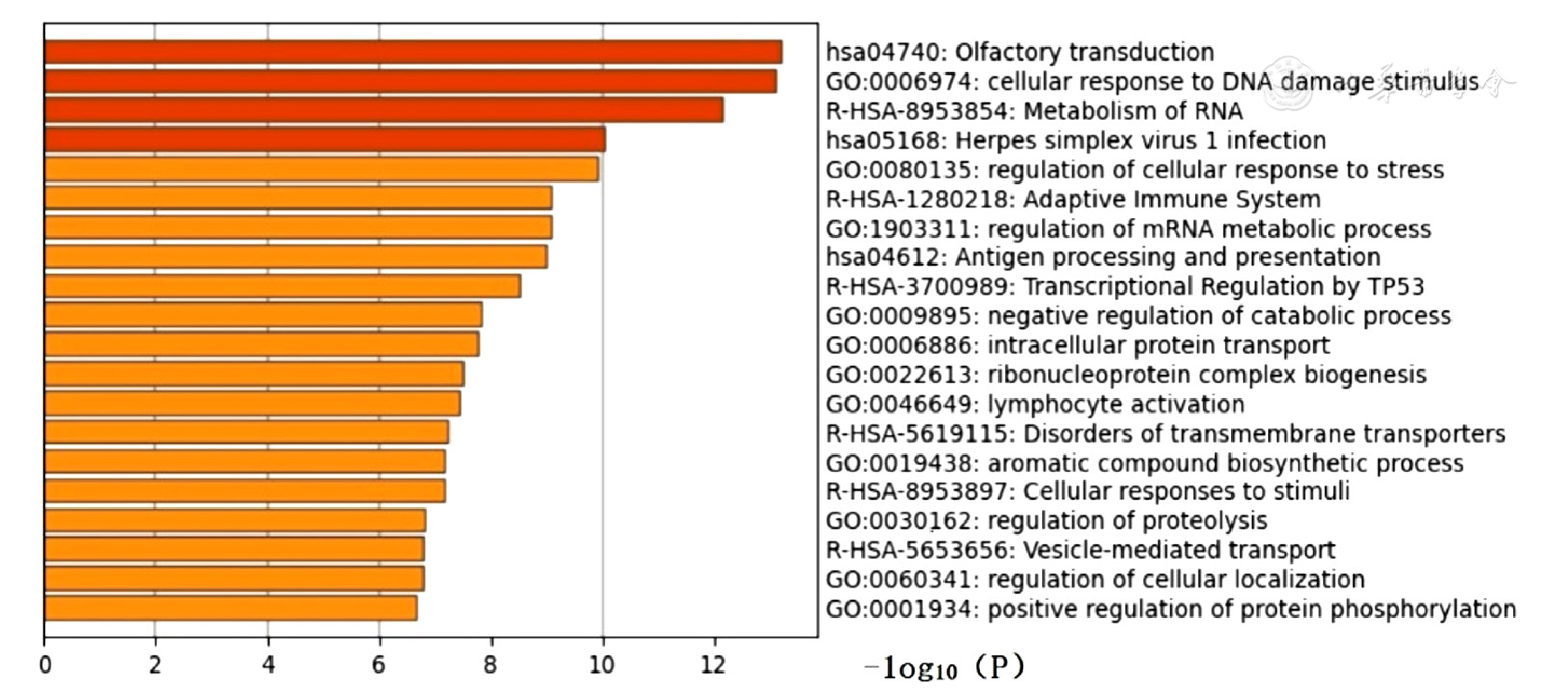
Figure 6 Metascape enrichment analysis of differentially expressed genes in epicardial adipose tissue
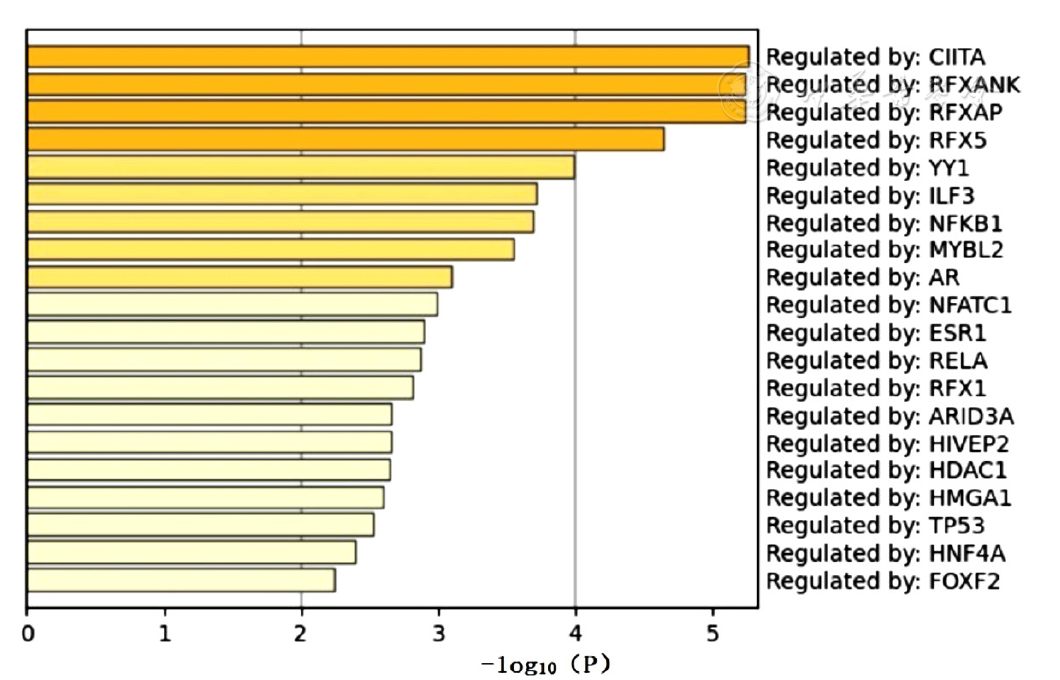
Figure 7 TRRUST database-based analysis of differentially expressed genes in epicardial adipose tissue to predict transcription factors
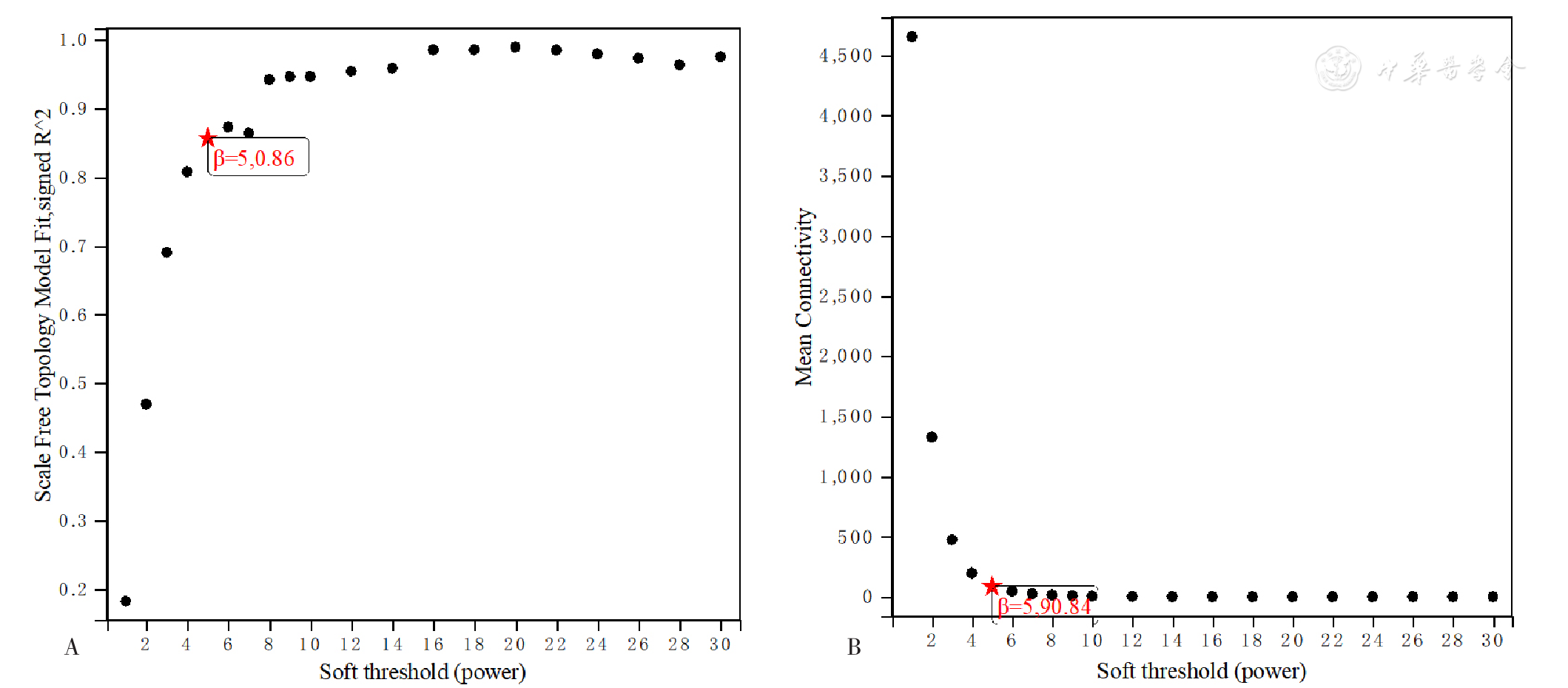
Figure 8 Network topology for soft thresholding powers
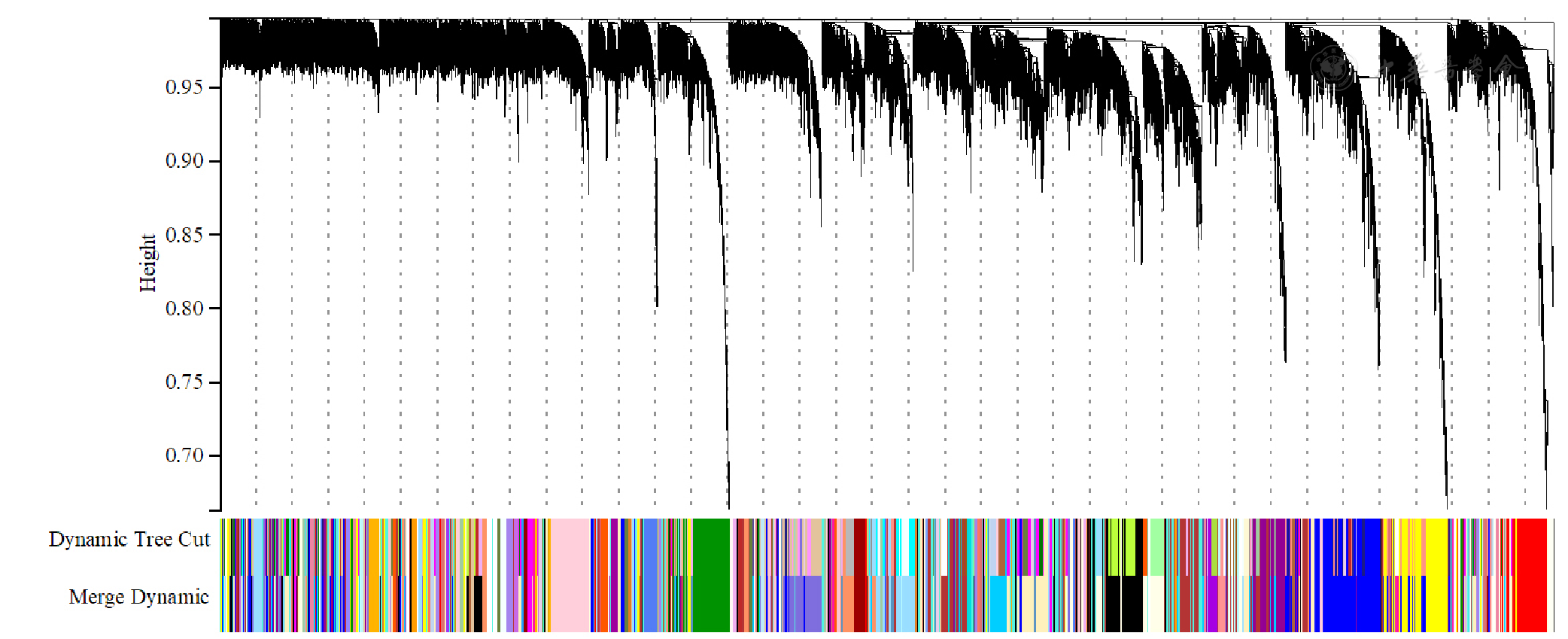
Figure 9 Dendrogram of gene clustering
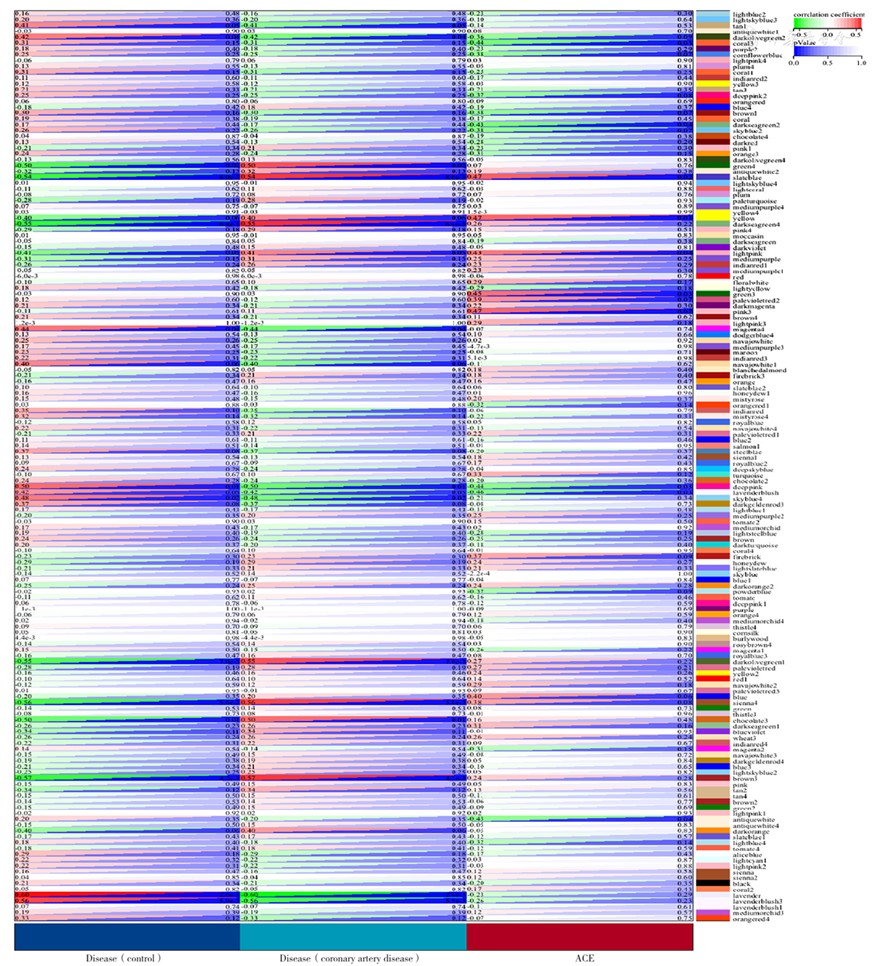
Figure 10 Correlation between gene modules and clinical information
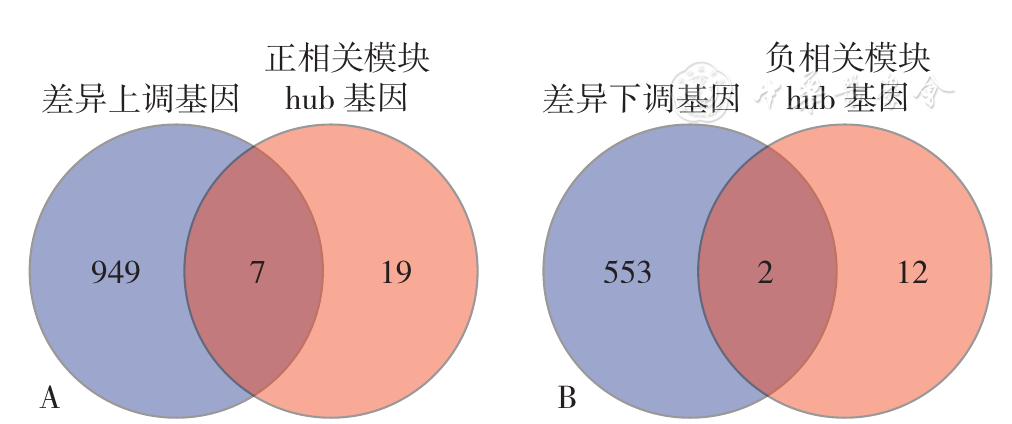
Figure 11 Venn diagram of differentially expressed genes and hub genes in the module
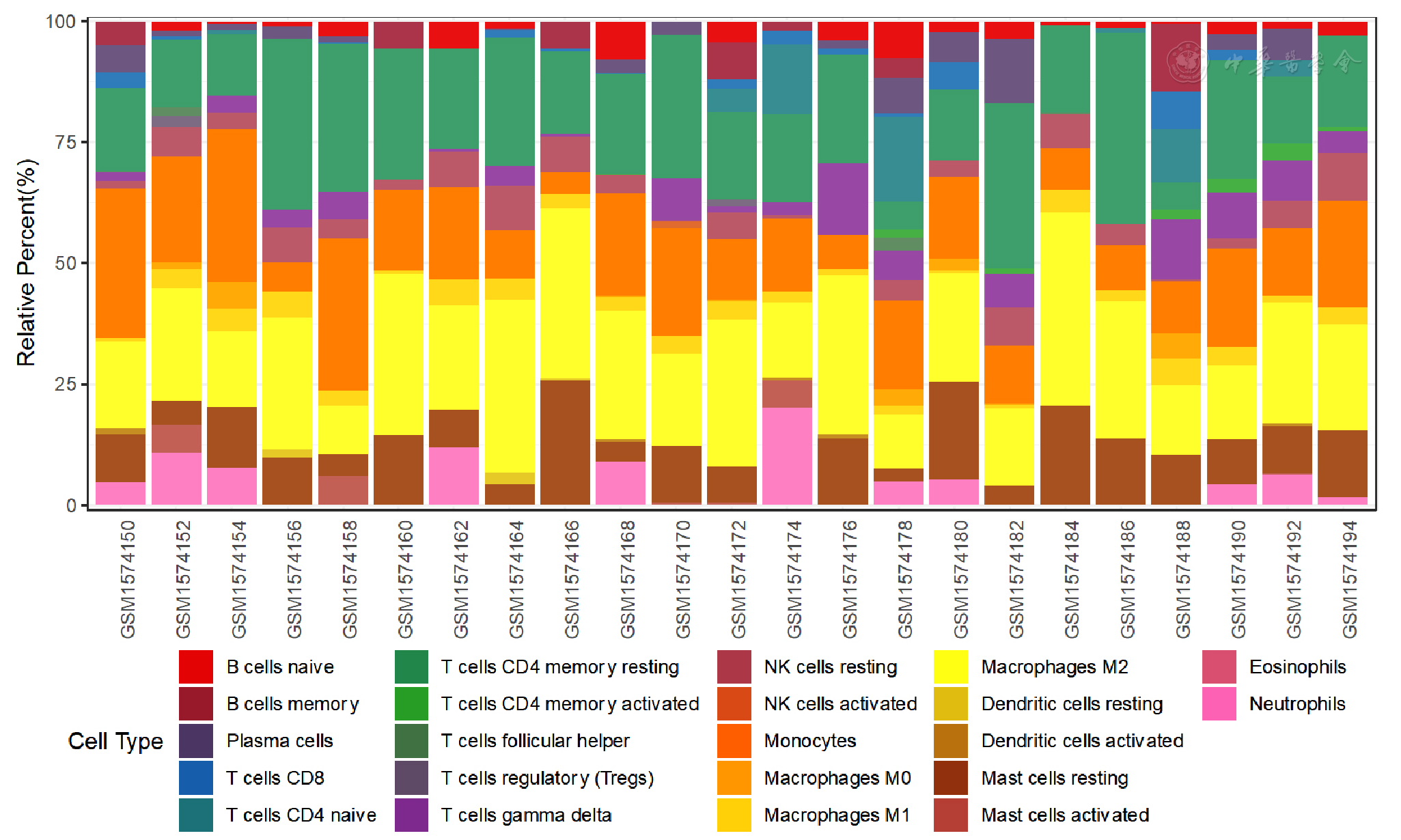
Figure 12 Bar graph of immune cell infiltration in epicardial adipose tissue between CAD patients and controls included in dataset GSE64554
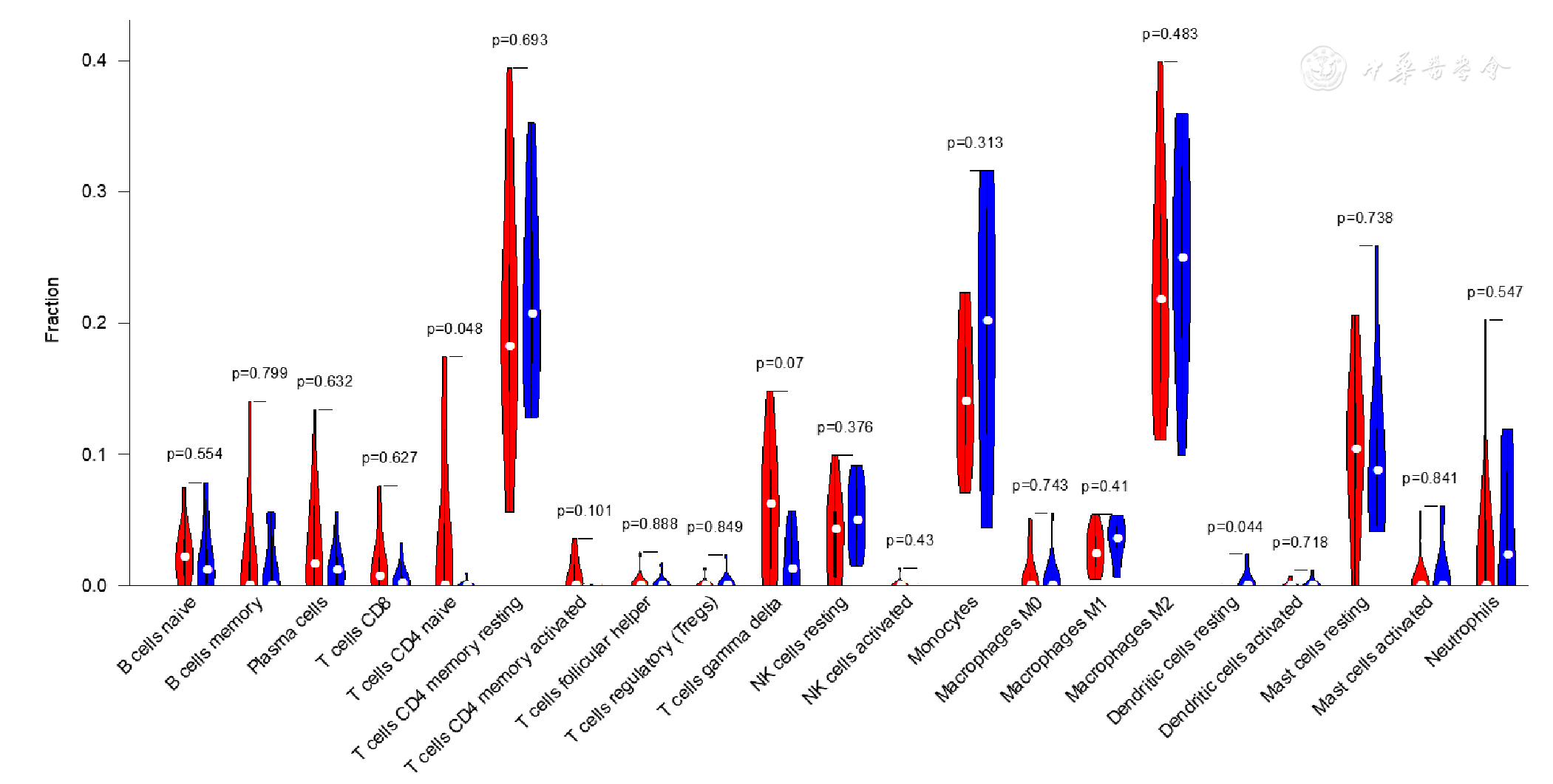
Figure 13 Violin plot of immune cell infiltration in epicardial adipose tissue between CAD patients and controls included in dataset GSE64554
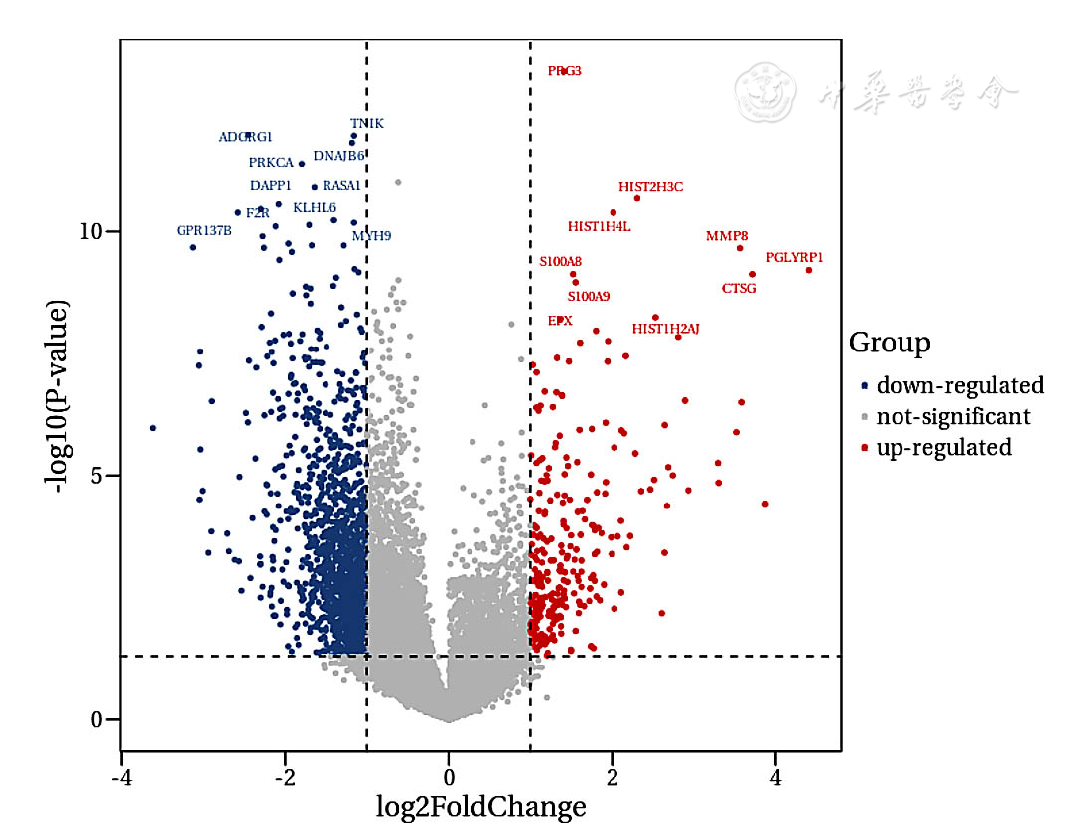
Figure 14 Volcano plot of differentially expressed genes in exosomes between CAD patients and heathy controls
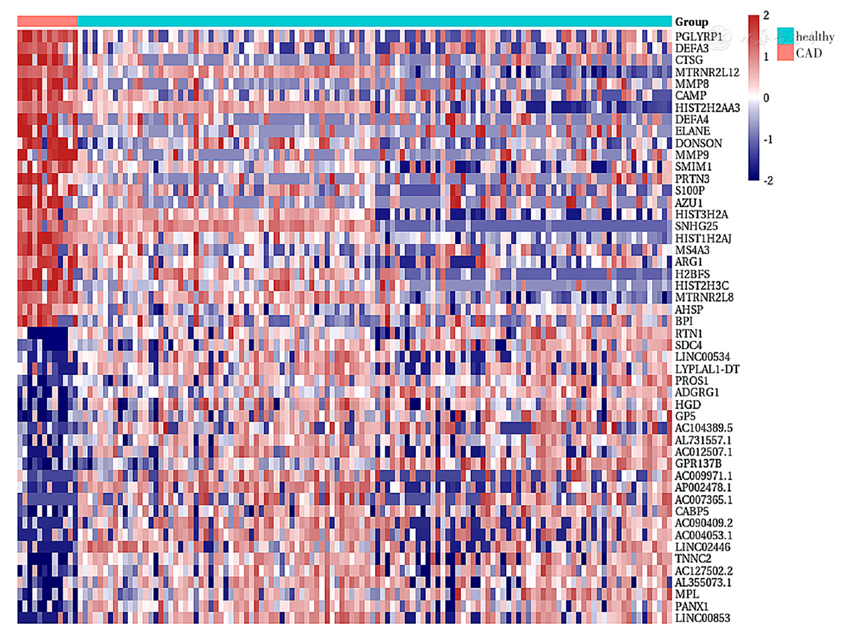
Figure 15 Heatmap of differentially expressed genes in exosomes between CAD patients and heathy controls
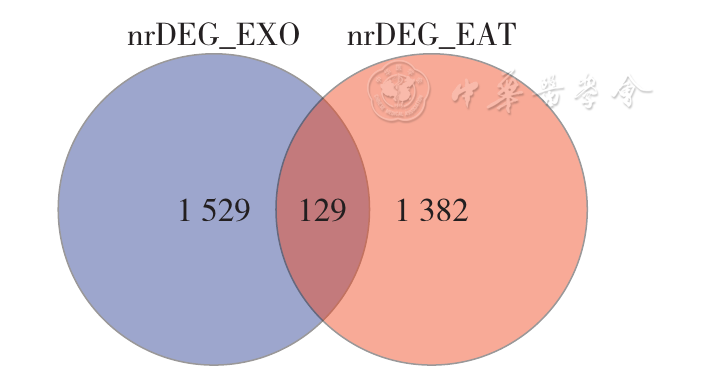
Figure 16 Venn diagram of differentially expressed genes in epicardial adipose tissue and exosomes from CAD patients
| 组别 | 样本数 | BPI | BIRC5 | CXCL12 | RNASE1 | F2R |
|---|---|---|---|---|---|---|
| CAD组 | 10 | 2.14±0.32 | 1.98±0.23 | 2.91±0.89 | 4.47±1.56 | 0.45±0.06 |
| 健康对照组 | 10 | 0.99±0.17 | 1.02±0.11 | 1.14±0.44 | 1.28±0.56 | 1.00±0.12 |
| t值 | -9.945 | -12.070 | -5.630 | -6.001 | 12.637 | |
| P值 | 0.011 | 0.010 | <0.001 | 0.009 | <0.001 |
Table 2 Differentially expressed genes in exosomes between CAD patients and healthy controls
| 组别 | 样本数 | BPI | BIRC5 | CXCL12 | RNASE1 | F2R |
|---|---|---|---|---|---|---|
| CAD组 | 10 | 2.14±0.32 | 1.98±0.23 | 2.91±0.89 | 4.47±1.56 | 0.45±0.06 |
| 健康对照组 | 10 | 0.99±0.17 | 1.02±0.11 | 1.14±0.44 | 1.28±0.56 | 1.00±0.12 |
| t值 | -9.945 | -12.070 | -5.630 | -6.001 | 12.637 | |
| P值 | 0.011 | 0.010 | <0.001 | 0.009 | <0.001 |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||